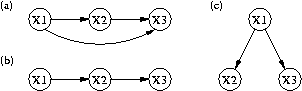
Figure 1. Three qualitative models of gene regulation that relate expression variables X1, X2, and X3.
Although biologists understand the basic mechanisms through which DNA produces biochemical behavior, they have not yet determined most of the regulatory networks that control the degree to which each gene is expressed. In collaboration with researchers from the Center for Computational Astrobiology at NASA Ames Research Center and the Department of Plant Biology at the Carnegie Institution of Washington, we are developing computational tools to help represent, utilize, and discover such regulatory networks. Our approach combines ideas from qualitative physics, causal models, theory revision, and statistical analysis.
One contribution of our work involves the representation of regulatory models in ways that make contact with biologists' ways of thinking while still being interpretable by computers. To this end, we have focused on qualitative causal models, which describe relations between quantitative variables (such as gene expression levels) but only specify the direction of the causal influence and its sign, not the functional form or parameters. Qualitative causal models provide a useful middle ground between quantitative models, which require many observations to fit parameters, and Bayesian networks, which, as typically used, rely on methods for discretizing continuous variables. We have also extended our qualitative formalism to support models with time-delayed effects.
A second contribution concerns computational methods for making predictions from such qualitative models. Expression data are quantitative in nature, but a qualitative model cannot predict continuous values. However, they can predict which variables should be correlated, along with the signs of those correlations. With additional assumptions about linearity, they can also predict relations among partial correlations, as noted by the developers of Tetrad. At an intuitive level, a partial correlation between two variables corresponds to the correlation between those variables when one controls for the effect of other variables. For example, consider the three qualitative causal models in Figure 1, which imply correlations between the three expression variables X1, X2, and X3. However, model (b) implies that the partial correlation between X1 and X3 given X2 is zero, whereas the others do not make this prediction.
|
|
| Figure 1. Three qualitative models of gene regulation that relate expression variables X1, X2, and X3. |
Yet another contribution involves computational methods for revising models of gene regulation in response to expression data. The basic technique carries out a heuristic search through the space of qualitative causal models, starting from an initial model provided by a biologist. Revision operators include adding a new link, removing a link, and reversing the direction of a link. The evaluation metric gives higher scores to candidates that predict more of the observed relations among partial correlations and that predict fewer of those not observed. Once the causal structure has been determined, a separate stage selects a sign for each link, in this case preferring models that better predict the signs of simple correlations. For models with time delays, the method carries out another level of search to select the best delay for each causal link.
A fourth contribution extends this approach to let it discover models of gene regulation from small data sets. Despite the common rhetoric about how much data has been made available by microarray technology, in most cases relatively few samples are available. We have responded to this challenge in two ways. First, we incorporate biological knowledge where possible to constrain the search for candidate models. Starting the search from an initial model provides one form of knowledge, but we also place constraints on the links considered during the revision process. Second, we utilize a variant of bootstrap sampling, in which we run the revision algorithm many times on slightly different versions of the data, then implement only those link changes that appear in most revised models. Both techniques should reduce variance and improve behavior on the small data sets we have available.
Finally, we are developing an interactive environment that will let biologists state qualitative models, encode background knowledge, evaluate models in the context of this knowledge and expression data, and suggest model revisions that are biologically plausible. We believe that biologists will find such an interactive environment more attractive than more automated methods that leave no room for input from the scientist. As part of this framework, we are developing tools that will let users visualize their models and expression data's relation to these models.
In our ongoing research, we are applying these computational methods to new data on gene expression from a number of sources. In addition, we are extending our methods to handle expression data from gene knockout experiments, to support nonlinear causal influences, to encode more abstract biological concepts like signaling chains, and to handle mechanistic models that describe biological processes in more detail. Our approach to modeling gene regulation has much in common with that taken by Clark Glymour and his colleagues, but differs in its focus on qualitative causal models, revision of those models, and interactive modeling environments.
This work has been supported by Grants NCC 2-1202, NCC 2-5471, and NCC 2-1335 from NASA Ames Research Center, and by NTT Communication Science Laboratories, Nippon Telegraph and Telephone Corporation. Researchers involved in the effort include Stephen Bay, Lonnie Chrisman, Pat Langley, Andrew Pohorille, Kazumi Saito, and Jeff Shrager.
Saito, K., George, D., Bay, S., & Shrager, J. (2003). Inducing biological models from temporal gene expression data. Proceedings of the Sixth International Conference on Discovery Systems. Sapporo, Japan.
Bay, S. D., Chrisman, L., Pohorille, A., & Shrager, J. (2003). Temporal aggregation bias and inference of causal regulatory networks. Proceedings of the IJCAI-2003 Workshop on Learning Graphical Models for Computational Genomics.
For more information, send electronic mail to langley@isle.org
 |
© 1997 Institute for the Study of Learning and Expertise. All rights reserved. |